
Compliance with Risk Minimisation Strategies – Expectations from Regulatory Authorities
Pharmacovigilance, the science of monitoring, assessing, and preventing adverse effects of medications, plays a crucial role in ensuring patient safety. A way regulatory authorities can ensure this is to request marketing authorisation holders (MAHs) to implement Risk Minimisation Strategies to manage and mitigate the risks associated with certain marketed drugs. Routine Risk Minimisation Measures (RMMs) are applicable to all medicinal products and involve the use of tools such as the Summary of Product Characteristics (SmPC), the package leaflet, the pack size and design, the labelling and the legal status of the product. These tools can be utilised to adequately address the safety concerns of a product. Where this may not be the case, additional Risk Minimisation Measures (aRMMs) may be implemented. Examples of this are the Pregnancy Prevention Programme (PPP) set up by the UK Medicines and Healthcare products Regulatory Agency (MHRA), and the Risk Evaluation and Mitigation Strategies (REMS) programme introduced US Food and Drug Administration (FDA). Although these programmes are comprised of different elements and are country-specific, they both have the common goal of minimising known potential risks to the patient, and helping healthcare professionals (HCPs) and patients make informed decisions. However, ensuring compliance with these strategies can present a significant challenge for pharmaceutical companies. At the DIA Global Annual Conference 2023, Sarah Gomersal (Pharmacovigilance Inspector, MHRA) and Haley Seymour (Compliance Officer, FDA) presented a session on RMMs. With their expertise, they gave an overview of the measures involved in implementing risk minimisation strategies, the expectations from the MHRA and FDA during risk minimisation inspections and provided examples of past inspection findings.
Additional Risk Minimisation Measures:
For UK-authorised medicinal products, aRMMs include the use of educational materials and complex aRMMs which comprise of controlled access/distribution programmes and PPPs. For products that require complex aRMMs, the MHRA will assign someone to work closely with MAH to manage the implementation processes. A complex aRMM programme developed by the MHRA is the PPP for teratogenic products. The PPP is intended to protect women of childbearing potential by preventing or controlling access of certain medications in these populations. To ensure this, there are requirements imposed upon several different involved parties.
1) Healthcare Professionals:
· Read required risk materials pertaining to the product
· Must be registered with PPP before they can provide the product
2) Product prescribers and dispensers:
· Ensure PPP requirements are adhered to with patient before providing the product
· Must enter data into PPP platform/system when product has been provided
3) Marketing Authorisation Holders:
· Review data from PPP platform/system to assess adherence
· Take appropriate action if non-adherence is identified
· Report data to MHRA
Between 2021 – 2023, a total of 50 Good Pharmacovigilance Practice (GPvP) Inspections were conducted by the MHRA, 11 of which were triggered aRMM inspections.
MHRA expectations of inspecting complex aRMMs:
· Written procedures e.g. Standard Operating Procedures (SOPs), Work Instructions (WIs), Templates
· Contracts and agreements between partners and vendors
· Trained personnel
· Computer systems and software have been validated
· Clear oversight of activities
· Adherence to the requirements of aRMMs
MHRA findings of PPP inspections:
· Computer system validation: Bespoke platforms created to support the PPP were not fully validated and there was no evidence of testing.
· Resources and processes: The MAH forecasted high volume of activities but resources were insufficient.
· PPP platform: Pages where HCPs entered data did not align with the requirements agreed with MHRA. Therefore, HCPs incorrectly entered information onto platform resulting in errors in wording and mandatory fields.
· HCPs were asked to only enter compliant data onto PPP platform – non-compliances were not recorded and identified.
· PPP platform generated inaccurate audit report.
· Lack of detail in supporting documents for PPP platform.
· Gaps in contracts and agreements.
The following are points to consider when implementing a complex aRMM, as advised by the MHRA:
· When developing complex aRMMs, the MHRA will work closely with the MAH to agree upon the unique requirements specific to the product – the MAH must ensure these requirements are met.
· Bespoke platforms should be accurate in reporting – MAH should maintain oversight of system performance and ensure the user functionality specifications are met.
· The MAH should continue to assess and monitor the performance of the aRMM after implementation and identify any issues.
· Ongoing support and cooperation with senior management at the MHRA for the aRMM is vital.
Risk Evaluation and Mitigation Strategies:
A REM is a risk management plan implemented for medicinal products authorised in the US. This plan uses risk minimisation strategies that go beyond product labelling to ensure that the benefits of the drug outweigh the risks. The FDA has also implemented a shared system REMS which allows multiple MAHs of a product to share a REM for that drug. This sharing increases transparency between MAHs by enabling access to larger amounts of relevant safety data and reduces burden on the FDA in handling multiple REMS of one product.
Each REMS programme is unique and is required only for some products. To determine which products require REMs, the FDA will consider:
· Estimated size of population likely to use the drug
· Seriousness of the disease or condition treated
· Expected benefit of the drug with respect to the disease or condition
· Expected o actual duration of treatment
· Seriousness of any known or potential adverse events
· Whether the drug is a new molecular entity (NME)
Possible elements of a REMS include:
· Medication guide or patient package insert
· Communication plan for HCPs
· Certain packaging and safe disposal technologies for drugs that pose a serious risk of abuse or overdose
· Elements to Assure Safe Use (ETASU) – most important element for drugs with highest risk
· Implementation system
· Timetable for periodic submission of assessments
There are many different parties involved in the monitoring, distribution and administration of a drug. Therefore, in the implementation and management of a REMS, there are many different responsibilities involved:
1) Drug manufacturers:
o Develop and implement REMS
o Assess REMS effectiveness in meeting its risk minimisation goals
o Submit reports of the REMS assessments to FDA at specific times
2) Healthcare providers:
o Enrolment and registration into the REMS
o Completion of REMS-specific training
o Enrolment of patients into the REMS registry
o Documentation of safe use conditions
3) Pharmacies:
o Have certification to dispense the medication
o Completion of REMS-specific training
o Counsel patients
o Provide educational materials or medication guide to patients
4) Patients:
o Be informed of symptoms to watch for and report
o Sign acknowledgement forms prior to starting medication
o Lab testing
o Enrol in regular monitoring
5) FDA:
o Determine if REMS is needed
o Review proposed REMS
o Review assessment reports by manufacturers
o Determines if REMS meets risk minimisation goals
o Can request REMS modification
Inspections of REMS compliance are conducted by the Office of Compliance, Postmarketing Safety Branch. They will determine the need for REMS inspections using a risk-based approach using the following considerations:
· REMS with ETASUs that have never been inspected
· REMS with ETASUs with documented issues from the previous Inspection
· REMS with ETASUs that were modified since the last inspection
· REMS with Communication Plans that have never been inspected
· REMS with any other issues that have been documented by FDA offices Investigation includes processes (SOPs, vendor contracts etc.), adherence to processes and validation of software
In a REMS inspection, FDA investigators will review:
· Processes including SOPs, contracts, employee training
· Validation of computer systems and call centres
· Adherence to SOPs, REMS documents and REMS requirements for all stakeholders
· Oversight of vendor’s responsibilities
Example findings:
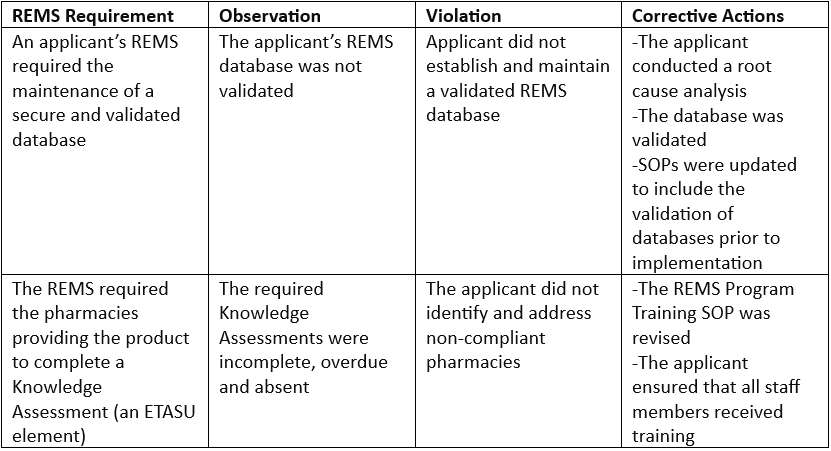
Best practices for inspections as recommended by the FDA:
· Ensure key documents and records are easily accessible, including:
o Procedures related to REMS activities
o Quality system, including a quality manual if applicable
o Organizational charts and tables
o Job/role descriptions and training protocols/records
· Ensure procedures are clear and concise
· Ensure issues since the last inspection are addressed and documented
· Provide documentation of corrective action
Drive Phase PV is a pharmacovigilance consultancy and provider of pharmacovigilance services, supporting patient safety throughout the life-cycle of our clients' products. Contact us to discuss assistance with your risk minimisation requirements..